Computing heritability
If you have missed the page to fit a simple univariate model in MCMCglmm, click here (Yes, you were supposed to click on MCMCglmm in the menu on the left to get to the first page)
Here we show how to compute narrow-sense heritability from a simple linear animal model. Narrow-sense heritability, or heritability for short, can be defined as the ratio of additive genetic variance over phenotypic variance. In our case, we have modelled only the mean, the additive genetic variance and the residual variance, so heritability is: $ h^2 = V_A / V_P = V_A / (V_A + V_R)$.
We still use the gryphon dataset with birth_weight as the response, and MCMCglmm.
phenotypicdata <- read.csv("data/gryphon.csv")
pedigreedata <- read.csv("data/gryphonped.csv")
library(MCMCglmm)
inverseAmatrix <- inverseA(pedigree = pedigreedata)$Ainv
We re-run the model we used previously:
model1.2 <- MCMCglmm(birth_weight ~ 1, #Response and Fixed effect formula
random = ~id, # Random effect formula
ginverse = list(id = inverseAmatrix), # correlations among random effect levels (here breeding values)
data = phenotypicdata, # data set
burnin = 10000, nitt = 30000, thin = 20) # run the model for longer compare to the default
summary(model1.2)
##
## Iterations = 10001:29981
## Thinning interval = 20
## Sample size = 1000
##
## DIC: 3912.137
##
## G-structure: ~id
##
## post.mean l-95% CI u-95% CI eff.samp
## id 3.406 2.227 4.668 379.1
##
## R-structure: ~units
##
## post.mean l-95% CI u-95% CI eff.samp
## units 3.851 2.886 4.91 444.5
##
## Location effects: birth_weight ~ 1
##
## post.mean l-95% CI u-95% CI eff.samp pMCMC
## (Intercept) 7.591 7.323 7.878 888.6 <0.001 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
One could get a rough calculation of heritability using the values in the summary, but it is much better to apply (i.e., integrate) the calculation on the full posterior distribution. It is actually quite simple in MCMCglmm and in general an advantage of Bayesian approaches.
We extract the vector of additive genetic variance posterior values (there are 1000 of them), stored in $VCV[,"id"] and the vector of residual variance posterior values (also 1000 of them), stored in $VCV[,"units"] and compute the 1000 posterior values of heritabilities as:
h2_full_posterior <- model1.2$VCV[, "id"] / (model1.2$VCV[, "id"] + model1.2$VCV[, "units"])
We can then look at the trace and distribution of heritability:
plot(h2_full_posterior)
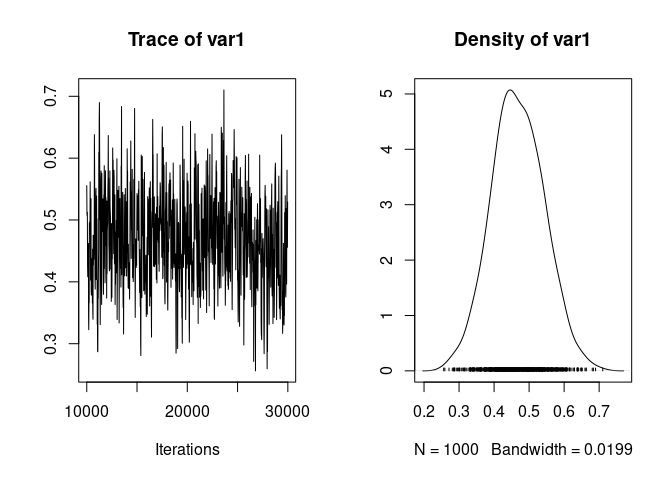
at point estimates:
mean(h2_full_posterior)
## [1] 0.4681129
median(h2_full_posterior)
## [1] 0.4645503
posterior.mode(h2_full_posterior)
## var1
## 0.4945104
and at credible intervals:
HPDinterval(h2_full_posterior) # default 95% interval
## lower upper
## var1 0.3280573 0.6111057
## attr(,"Probability")
## [1] 0.95
HPDinterval(h2_full_posterior, prob = 0.98) # another arbitrary interval with 98% probability
## lower upper
## var1 0.3002681 0.6515323
## attr(,"Probability")
## [1] 0.98
Feedback
Was this page helpful?
Glad to hear it! Please tell us how we can improve.
Sorry to hear that. Please tell us how we can improve.